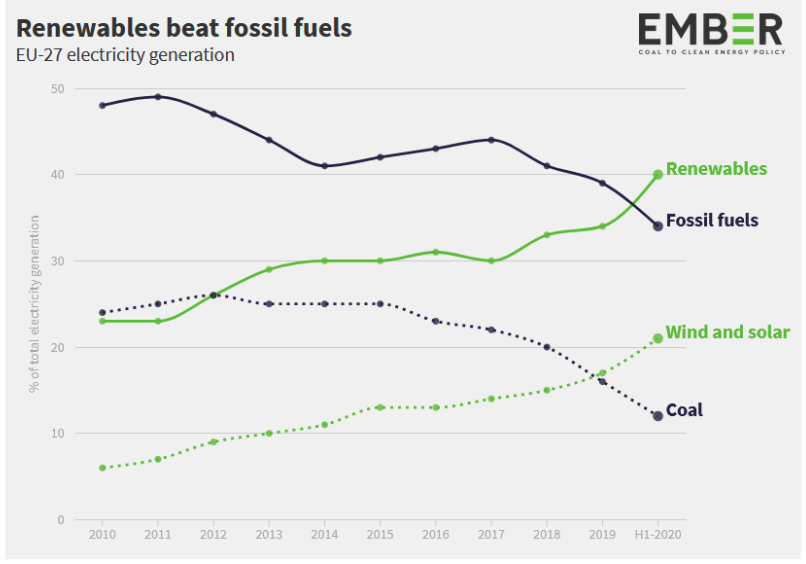
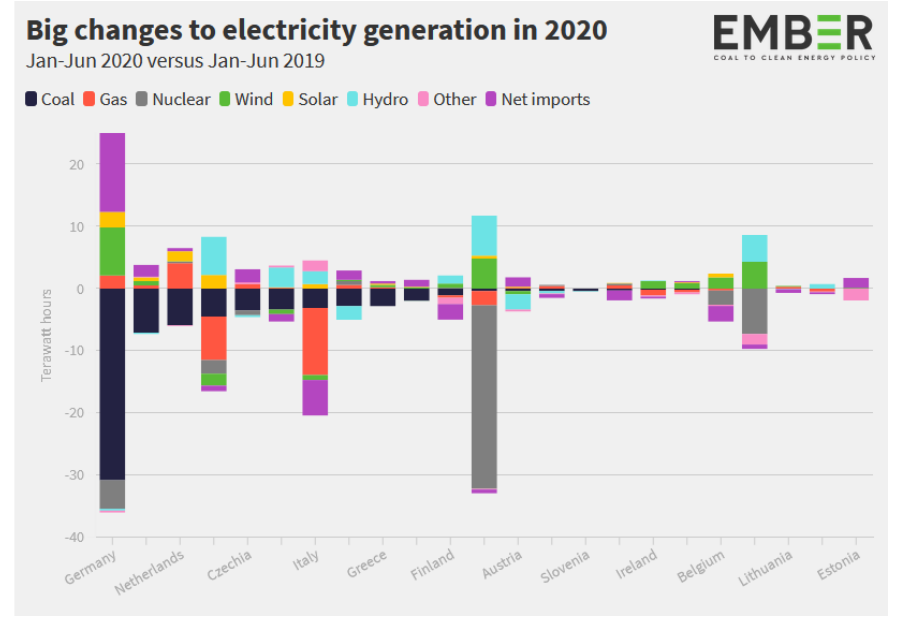
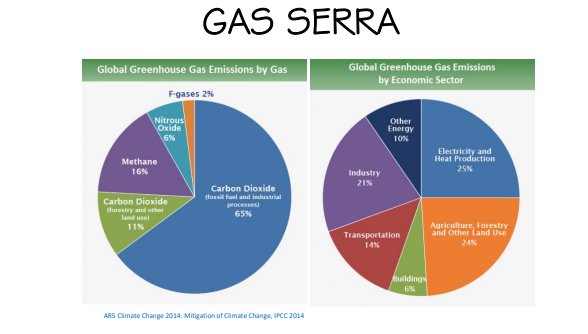
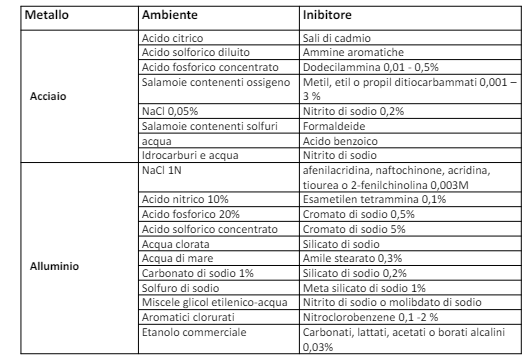
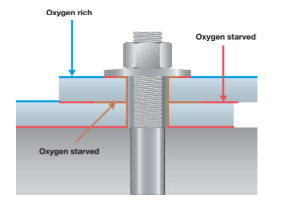
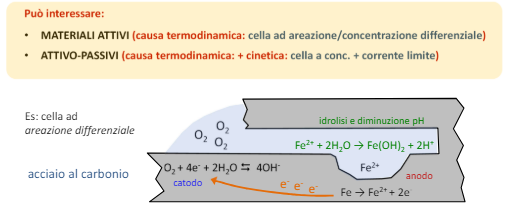
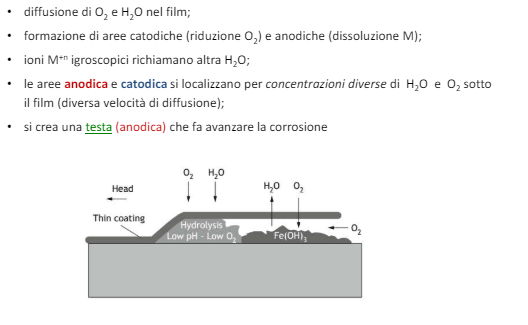
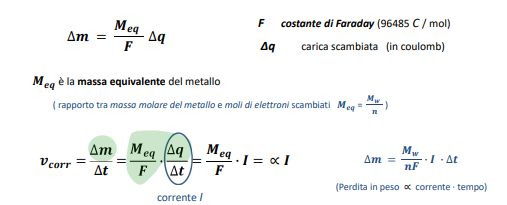
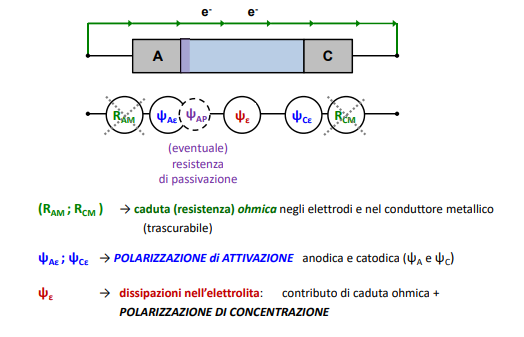
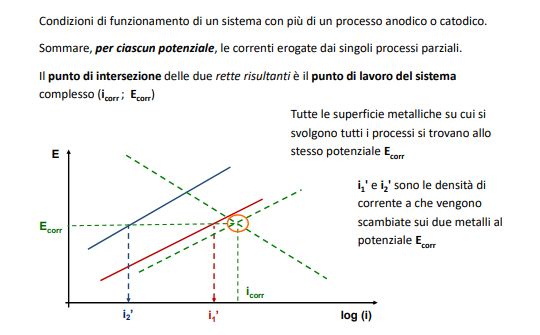
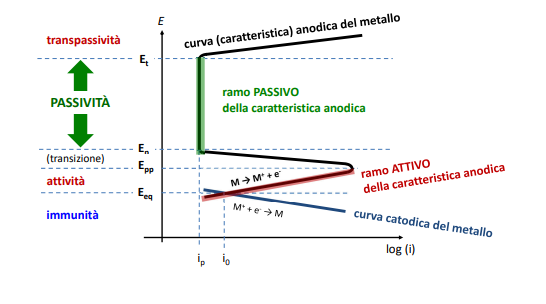
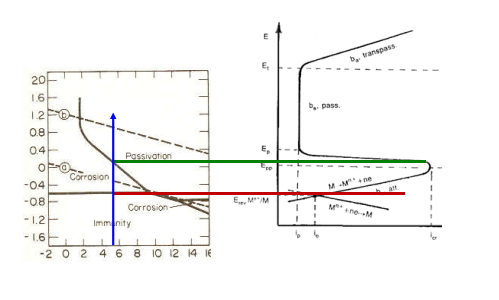
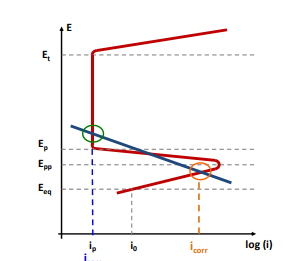
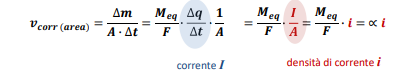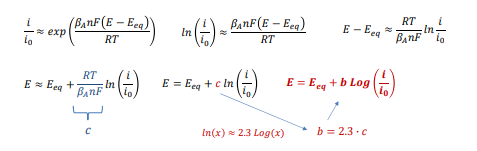Entriamo nel vivo dell’elettrotecnica con l’introduzione ai circuiti elettrici. Si intende come circuito un insieme di componenti el3ttrici collegati tra di loro. In questa prima parte vedremo la prima parte di teoria necessaria a comprenderli bene, nella seconda invece andremo direttamente a metterci mano.
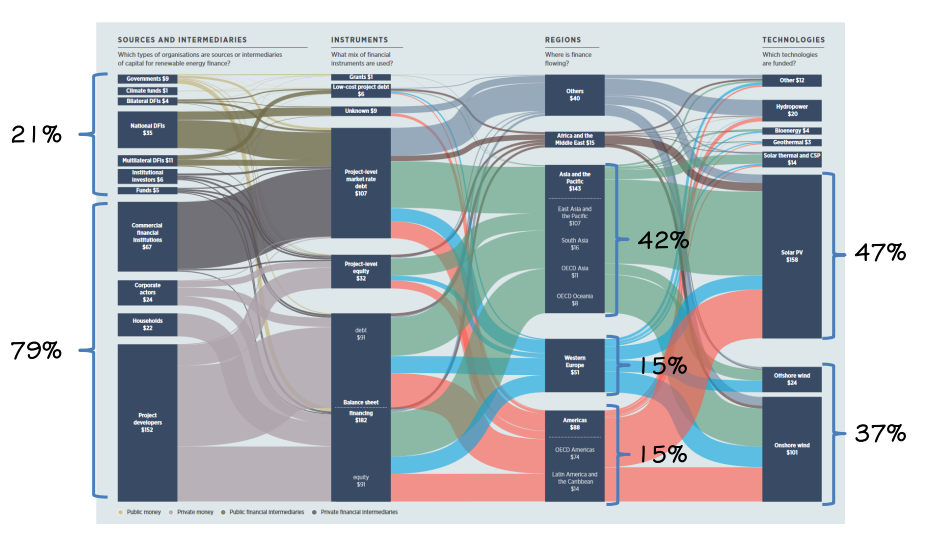
Iniziamo quindi dalla teoria dell’elettricità, ovvero:
Elettromagnetismo
L’elettromagnetismo è una branca della fisica che studia il comportamento e le influenze di cariche magnetiche sia tra di loro che con il mondo esterno ed è la scienza alla base di tutti i moderni dispositivi elettrici e elettronici e permette di pensare in modo ingegneristico a degli strumenti che permettano di utilizzare questo tipo di energia ai nostri scopi (muoverci, riscaldarci, illuminazione ecc…)
Quello che manca è la tecnica per poter sfruttare al meglio le conscienze prese dall’elettromagnetismo.
Infatti da questa scienza fisica si è poi divisa una scienza tecnica (l’elettrotecnica) che costruisce modelli matematici al fine di modellare e sviluppare dei modelli ideali e poi trasformarli in modelli reali permettendo quindi l’utilizzo della fisica dell’elettromagnetismo in campo ingegneristico e tecnico.
Questo accade analizzando i componenti come scatole nere che cambiano e modificano dei fattori.
Quindi gli scopi del corso sono quelli di prevedere come si comporta un circuito e poi poterne ideare di nuovi.
Esistono 2 tipi di dispositivi:
- elettrici: applicazioni dell’energia: generazione, trasporto e usufrutto dell’energia elettrica (esempio generatori, centrali, cablaggio e trasporto di potenza);
- elettronici: dispositivi per applicazioni informatiche (computer,telefoni ecc), strumenti per creare e processare informazioni (esempio radio, televisione, telefoni e pc)
Nella realtà si utilizzano dispositivi elettronici e per gestire energia elettrica e l’energia elettrica per alimentare dispositivi elettronici rendendo quindi le due branche strettamente interconnesse tra loro.
Esistono anche applicazioni più nuove di elettronica e elettricità come pannelli solari e auto elettriche, per la medicina ecc…
Teoria dei circuiti concentrati
La teoria dei circuiti concentrati è ricavata dalle equazioni di Maxwell che sono alla base dell’elettromagnetismo.
Questa teoria ha però dei limiti, infatti questo non entra direttamente nel circuito fisicamente ma vediamo i vari componenti come se fossero scatole nere senza interessarci di cosa avviene al loro interno.
Per poter trovare i limiti della teoria bisognerebbe vedere il tempo di transito (il tempo che ci mette l’elettrone a passare da una parte all’altra del componente) dell’elettrone:infatti se il tempo di transito dell’elettrone è molto minore del tempo che ci mette il dispositivo a variare il suo effetto nel sistema, allora il circuito si può considerare a parametri concentrati.
Noi possiamo dire che se il tempo di transito è molto piccolo rispetto all’intervallo possiamo considerare la corrente entrante e uscente uguale e usare la teoria dei parametri concentrati al posto delle leggi di Maxwell.
Carica elettrica
La carica elettrica è una grandezza fisica fondamentale ed è quella che genera la forza di attrazione o di repulsione tra due corpi caricati elettricamente rispettando la legge di Coulomb.
le cariche possono avere segni opposti (+ e -) e la carica fondamentale presa in considerazione è la carica degli elettroni (carica convenzionalmente negativa).
In caso di cariche di stesso segno esse si respingono mentre le cariche di segno opposto si attraggono.

La carica elettrica ha come unità di misura il Coulomb.
Un Coulomb è circa uguale 6.24 x 1018
Attenzione: la carica elettrica non si crea e non si distrugge ma circola all’interno del circuito (cioè gli elettroni si muovono lungo le varie componenti e i cavi).
Corrente elettrica
La corrente elettrica è definita come la quantità di carica che attraversa una determinata superficie in un determinato periodo di tempo
Convenzionalmente nonostante sia fisicamente sbagliato, il verso della corrente è determinato dal “movimento” delle cariche positive.(fisicamente sbagliato perchè a muoversi come corrente sono gli elettroni all’interno del materiale e non i nuclei degli atomi)

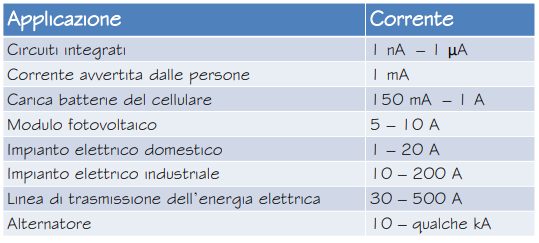
La corrente elettrica si misura in Ampere (C/s) tramite un Amperometro.
Tipicamente questo movimento di elettroni avviene nei metalli grazie alle bande di elettroni di conduzione esistenti a livello atomico.
I multipoli (dispositivi modellabili circuitalmente), gli elementi dei circuiti
In elettrotecnica si utilizzano vari “elementi” che permettono di svolgere alcune operazioni con i due dati fondamentali di elettrotecnica: la differenza di potenziale e la corrente.
Queste scatole comunicano tramite il mondo esterno con dei terminali o poli tramite i quali, a seconda della struttura del circuito all’interno della scatola, quest’ultimo riceve dati (sotto forma di intensità di corrente o differenza di potenziale) e emette dati (sempre sotto forma di corrente o DDP).
Sono esempi di terminali prese di corrente, resistenze, connettori delle auto elettriche caricatori dei cellulari, i piedini dei circuiti integrati o quelli dei processori.
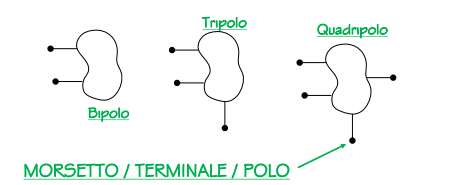
convenzioni
Nei circuiti la corrente non è determinata solo da un numero ma anche da un segno che ci fornisce il verso di scorrimento di questa all’interno di un circuito.
Convenzionalmente sarà per noi necessario fissare arbitrariamente e a priori un verso di corrente. Questo non influirà sulla validità dei calcoli ma eventualmente su alcuni segni.

Quindi :
- per ogni terminale si indica a priori un verso convenzionale della corrente,
- si eseguono i calcoli in riferimento a quella convenzione
- se i calcoli portano a un valore positivo BENE, altrimenti si intuisce che la corrente fluisce in senso opposto a quello adottato nella convenzione.
Campo elettrico
Come campo elettrico si intende la “sfera di influenza” di una carica e serve a spiegare l’interazione tra cariche elettriche a distanza, è quindi un campo di forze generato da una carica Q in cui inseriamo una carica q (detta di prova) per poterne vedere gli effetti.
Le formule del campo elettrico sono :


in cui la prima rappresenta vettorialmente l’intensità del vettore del campo elettrico (in poche parole è una sorta di ” gravità ”).
Campo elettrico
Ad una carica q posta in un campo E si trova un’energia associata alla singola carica che denotiamo con w.
l’energia per carica la chiameremo potenziale elettrico la cui formula sarà
V=wq
Questa energia (w) è possibile associarla per similitudine all’energia potenziale gravitazionale.
Tensione elettrica
Dati 2 punti a e b immersi in un campo elettrico, la tensione elettrica tra questi due punti è la differenza tra i potenziali di una stessa carica posizionata nei 2 punti. Viene infatti definita come

vab= -vba=V(a)-V(b)=w(a) – w(b)q=wq
La tensione elettrica o differenza di potenziale viene espressa in volt e viene misurata con uno strumento chiamato Voltmetro
1V=1jC
A titolo di richiamo la tensione dipende solo dai punti a e b senza considerare la carica spostata e il percorso svolto.
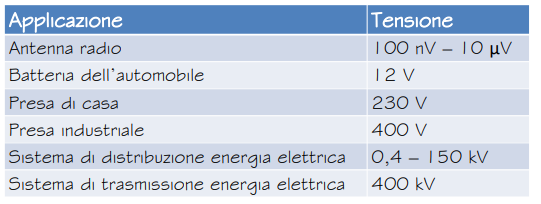
Polarità della tensione
Ai capi di una “scartola” il polo a potenziale maggiore è caratterizzato da un segno + (testa della freccia). Nei circuiti questo comporta il “senso di percorrenza” della corrente.
Convenzioni
Qui come per la corrente, a meno che non sia specificato a priori, sarà necessario anche in questo caso scegliere arbitrariamente la polarità e poi valutare a posteriori del calcolo se la nostra scelta è stata corretta e nel caso invertirla.
Forme d’onda
Nel corso delle lezioni incontreremo vari grafici e varie forme d’onda tra cui
(corrente continua, corrente alternata,segnali in PWM, carica e scarica di un condensatore, diodi, transistor, isteresi magnetica ecc…) Sarà necessario vedere quindi le variazioni al variare del tempo cosa accade.
Utilizzeremo molto la corrente alternata e la corrente continua.
qui riportiamo alcuni esempi
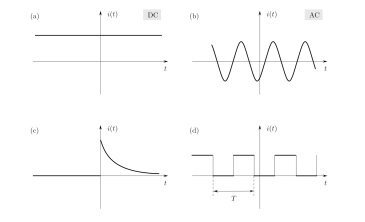
Esistono diverse fonti di energia a corrente continua: pannelli solari, batterie, convertitori a idrogeno, pannelli solari, dinamo ecc…
E utilizzatori in corrente continua come led, computer, server, telefoni ecc…
Così come esistono generatori a corrente alternata come alternatori, inverter generatori a gasolio ecc…
E utilizzatori a corrente alternata come motori trifase, motori bifase asincroni.
Potenza elettrica
La potenza elettrica è definita come la quantità di energia che il movimento delle cariche produce o perde passando da un potenziale all’altro (attraversamento del dipolo) in un certo periodo di tempo (in qualche modo riconducibile concettualmente all’energia cinetica e al lavoro gravitazionale)
Riducendo l’equazione è possibile ricavare varie formule per la potenza elettrica:

- p=v x i
- p=Ri2
- p=v2/R
vedremo più avanti il significato di R come resistenza.
La potenza inoltre si misura in watt tramite il Wattmetro (strumento).
Convenzioni per i circuiti
Anche in questo caso ci sono delle convenzioni per il segno e in particolare il verso della potenza scambiata al dipolo coincide con quello della corrente al polo positivo.
In poche parole se il verso della corrente al polo positivo allora la potenza sarà entrante (potenza assorbita).
Se in questo caso la corrente sarà negativa allora la potenza verrà negativa il che significa che fisicamente la potenza è uscente (potenza generata). portando quindi a 4 combinazioni:
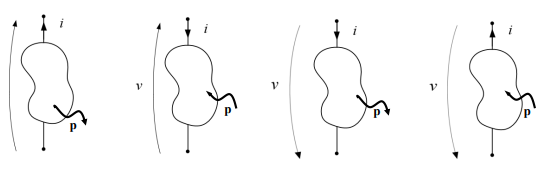
vediamo:
- caso 1 corrente uscente al polo positivo → potenza uscente
- caso 2 corrente entrante al polo positivo → potenza entrante
- caso 3 corrente uscente al polo positivo → potenza uscente
- caso 4 corrente entrante al polo positivo → potenza entrante
esempio 1.5 del Perfetti : calcolare la potenza assorbita da 1 e 2

esempio 1.6 del Perfetti : calcolare sempre potenza assorbita
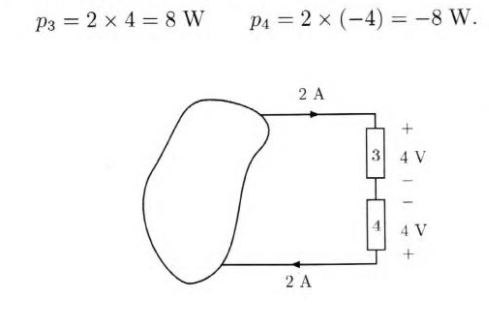
vediamo che una delle potenze assorbite è negativa; questo indica che in realtà la potenza non è assorbita ma emessa dal dipolo preso in considerazione.
Convenzioni per i circuiti
Da un dispositivo elettrico la potenza può essere assorbita o ceduta al circuito e questo determina la differenza tra utilizzatori e generatori.
Per il generatore si assegna verso positivo alla potenza ceduta, mentre per gli utilizzatori si assegna verso positivo alla potenza assorbita.

Vediamo per i generatori che corrente e DDP sono coordinati e la potenza ceduta ha valore positiva.
Per l’utilizzatore invece vediamo che la corrente e la DDP non sono coordinate e la potenza assorbita allora avrà valore positivo.

Il Wattora è la quantità di potenza assorbita o ceduta ogni lasso di tempo ed è una rappresentazione effettiva di energia (misurabile in joule).
1 Wattora= 3600 j
se la potenza non è costante allora si fa l’integrale della potenza nel tempo


Il costo el watt ora è influenzato da molti fattori, con riferimento al solo costo dell’energia questo è un mercato estremamente volatile e varia giornalmente di 土 0,08/0,1 €
Importante ai fini ambientali è il consumo medio annuo per una famiglia media si aggira intorno ai 2000 kWh
Esempi di calcolo di energia
Energia prodotta annualmente da una centrale

Convenzioni interne al corso
- Le grandezze elettriche variabili nel tempo si indicheranno con la lettera minuscola
esempio
i=i(t)
- Le grandezze elettriche fisse nel tempo saranno rappresentate con la lettera maiuscola
- ci saranno altre grandezze legate alle correnti alternate (fasori in numero complesso) che segneremo con una lettera maiuscola con un tratto sopra
esempio
Ī
Conduttori Ideali
(i fili che collegheranno i terminali tra una scatola e l’altra)
Si ipotizza per i conduttori ideali che i collegamenti tra un multipolo e l’altro siano equipotenziali, cioè all’interno dello stesso filo il potenziale è 0
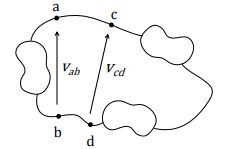
Sono quondi vere le seguenti affermazioni su questo circuito:
- Vab=Vcd
- Vac=0
- Va=Vc
- Vb=Vd
Questo implica che le variazioni di energia avviene solo all’interno dei multipoli
Reti o Circuiti elettrici
L’insieme di più multipoli collegati da conduttori ideali prende il nome di circuito e dato che i collegamenti sono equipotenziali e le variazioni di energia avvengono solo all’interno dei multipoli, questi circuiti vengono definiti a parametri concentrati.
Il modo in cui i vari elementi di un circuito sono collegati tra loro viene detta topologia del circuito.
Per la topologia non è importante la lunghezza del filo o la posizione nello spazio, ma solo e unicamente delle connessioni tra i multipoli.
Per esempio nell’immagine sotto i tre circuiti hanno stessa topologia perché i collegamenti sono sempre gli stessi
ESEMPIO 1A—1C ; 2C—2B

Lo studio dei circuiti
Nello studio del circuito i multipoli verranno visti come delle scatole nera al cui interno avvengono cose e che (come detto prima) interagiranno attraverso i loro terminali tramite variabili esterne che saranno tensioni e correnti.
L’analisi quindi del circuito si basa sulla volontà di ricavare tensioni e correnti ai vari terminali data la topologia degli elementi che lo compongono. In questa serie di relazioni saranno denominate relazioni caratteristiche e tramite le equazioni topologiche.
Leggi di Kirchhoff
Leggi di kirchhoff leggi fondamentali su cui si basa lo studio dei circuiti a parametri concentrati
Nodi,rami e maglie, nei circuiti
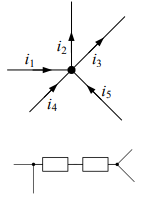
Un nodo è il punto di incontro di 3 o più poli, in poche parole un bivio di correnti.
Un ramo è l’insieme dei componenti compreso tra 2 nodi, cioè un gruppo di componenti connessi tra loro senza nodi intermedi.

Una maglia di un circuito e un insieme di rami in cui la corrente può scorrere in un percorso chiuso, Ogni maglia può contenere altre maglie al suo interno.
Prima legge di Kirchhoff
- 1 in un nodo la somma delle correnti è uguale a 0

i1+i4+i5=i3+i2
j=1nin=0 LKC
Esempio 1.2 perfetti

Esempio fatto in classe

perto dal nodo A che ha una sola incognita e scrivo l’equazione del solo nodo A per poter ricavare I2:
i7+i8=i6+i2 i7+i8-i6=i2 che in numeri (-1)+3-(1)=1
dal nodo B abbiamo equazioni
i5+i3+i2=i1+i4 i5+i3+i2-i4=i1 che in numeri 5+2+1-(-3)=11
Non sempre però è disponibile un “nodo di partenza” con una singola incognita; nel caso si costruisce un sistema a n equazioni in n incognite :
i7+i8=i6+i2 equazione del nodo A
i5+i3+i2=i1+i4 equazione del nodo B
Seconda legge di Kirchhoff
- 2 in una maglia la somma delle tensioni è uguale a 0
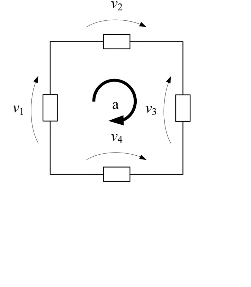
v1+v2+v3+v4=0
j=1nvj=0 LKT
Esempio 1.3 del Perfetti

In questo caso bisogna scegliere un senso di percorrenza della maglia per poter assegnare il segno alle varie DDP della maglia e assegneremo le frecce concordi al senso di percorrenza con segno positivo e quelle contrarie con segno negativo.
Per esempio se prendiamo il senso orario otterremo
4+10-v-12=0
mentre se prendiamo il senso antiorario otterremo
-4-10+v+12=0
Il risultato indiscutibilmente non cambia
Per le leggi di kirchhoff in poche parole quello che entra deve anche uscire
Esempio 1.5 del Perfetti
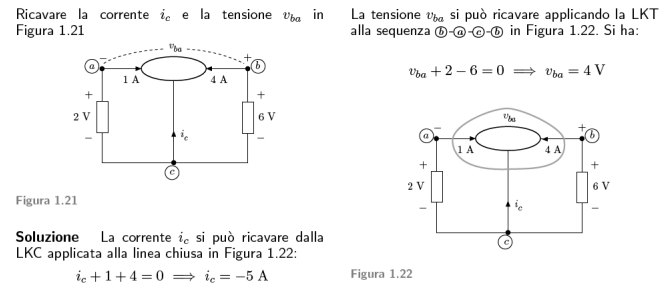
Equazioni di un circuito
Le leggi di Kirchhoff dipendono dalla topologia del circuito non dagli elementi che compongono il circuito.
Supponiamo di avere un circuito con n nodi e l lati
- applicando LKC otteniamo n-1 equazioni indipendenti
- applicando LKT otteniamo l-n+1 equazioni indipendenti
Quindi dalla sola topologia del circuito otteniamo [(N-l) +(L-N+I)]= L equazioni indipendenti che denotiamo come equazioni topologiche che però non sono sufficienti a risolvere il circuito, infatti per risolvere il circuito avremmo bisogno di 2L equazioni date dalle L tensioni + L correnti
Per ottenere le altre equazioni bisogna fare ricorso alle equazioni costitutive ( dette anche relazioni caratteristiche) degli elementi che lo compongono.
Conservazione della potenza istantanea

La somma algebrica delle potenze assorbite (o generate) da tutti gli elementi di un circuito è nulla in ogni istante. Questo significa in termini fisici che in un circuito ideale la potenza generata viene anche completamente assorbita.
Consideriamo due dipoli a e b collegati tra loro da conduttori ideali La potenza assorbita dal dipolo a e uguale a Pa=vi mentre la potenza erogata da b è Pb=-vi;
La somma algebrica delle potenze assorbite da tutti gli elementi di un circuito è nulla in ogni istante; in un circuito quindi la potenza generata dai generatori è uguale a quella assorbita dagli utilizzatori; ciò è valido solo per collegamenti con conduttori ideali.
Esempio 1.10 ( CAPIRE LA POTENZA ASSORBITA DAI BIPOLI)
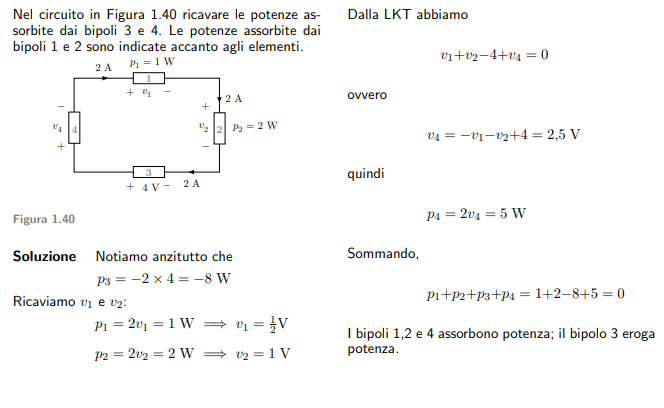
Per rimanere aggiornato clicca qui!!
Mentre per altri articoli di elettrotecnica clicca qui.